This cell adds /home/docs/checkouts/readthedocs.org/user_builds/wsu-phys-581-computation/checkouts/latest/src to your path, and contains some definitions for equations and some CSS for styling the notebook. If things look a bit strange, please try the following:
- Choose "Trust Notebook" from the "File" menu.
- Re-execute this cell.
- Reload the notebook.
Molecular Dynamics#
Another way to simulate material properties is with molecular dynamics (MD). This simply uses Newton’s laws to track the positions \(\vect{r}_{i}\) and velocities \(\vect{v}_{i} = \dot{\vect{r}}_{i}\) of many particles:
What makes MD simulations difficult is calculating the force \(\vect{F}_{i}\), which may depend on many or all of the other particles. For example, if the particles interact through a potential \(V(r)\), then we have formally
Details: Getting the right sign.
This comes from
If \(V(r)\) is repulsive, \(V'(r)\) will be negative, meaning that \(\vect{F}_{i}\) will point in the direction of \(\vect{r}_{ij}\), i.e., from \(\vect{r}_{i}\) (tip) to \(\vect{r}_{j}\) (tail) – away from the particle as expected for a repulsive potential.
Naïvely, this requires summing over all the particles – \(O(N)\) operations – for every particle, leading to a computational complexity of \(O(N^2)\). Part of the art of MD is using clever approximations to reduce this complexity. See [Griebel et al., 2007] for details.
Lennard-Jones Potential#
A common example is to use a Lennard-Jones potential:
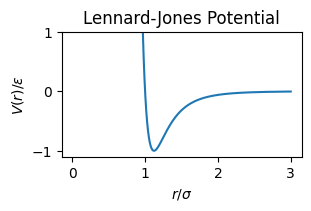
This has a minimum at \(r_\min = 2^{1/6}\sigma\) where \(V'(r_\min) = 0\), which can be used to create a stable crystalline solid, thereby allowing us to simulate the elastic properties of materials in extreme conditions (i.e., where they explode).
Time Evolution#
Care must be taken when evolving an MD simulation in time, otherwise, numerical errors will rapidly invalidate your simulation. Here we use the Velocity-Størmer-Verlet integration scheme
This requires storing the previous acceleration \(\vect{a}(t)\) with each step.
Here we present a simple simulation of a projectile impacting a slab (see Fig. 3.12 of
Griebel:2007). This is a simple code but runs slowly as it suffers from the \(O(N^2)\)
complexity (hence the small resolution shown here).
The philosophy here is to first code the simplest thing you can to make sure we understand the essential pieces of the algorithm, and that there are no roadblocks. Once you have this, it can form a test-bed for optimizing and exploring more complicated algorithms.
CPU times: user 10.9 s, sys: 4.99 s, total: 15.8 s
Wall time: 17 s